Rhizomelic chondrodysplasia punctata
| Rhizomelic chondrodysplasia punctata | |
|---|---|
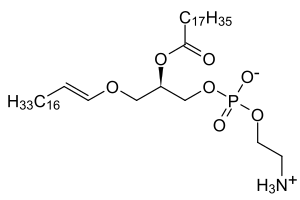 | |
| Low levels of plasmalogens is a characteristic of rhizomelic chondrodysplasia punctata. | |
| Symptoms | Alopecia, flat face[1] |
| Causes | PEX7 gene, GNPAT gene and AGPS gene mutations[2] |
| Diagnostic method | Clinical and radiologic finding[3] |
| Treatment | Physical therapy[4] |
Rhizomelic chondrodysplasia punctata is a rare developmental brain disorder characterized by systemic shortening of the proximal bones (i.e. rhizomelia), seizures, recurrent respiratory tract infections and congenital cataracts. The affected individuals have low levels of plasmalogens.[2]
Signs and symptoms
Rhizomelic chondrodysplasia punctata has the following symptoms:[4][1]
- Bilateral shortening of the femur
- Post-natal growth problems (deficiency)
- Cataracts
- Intellectual disability
- Possible seizures
- Possible infections of respiratory tract
Genetics
This condition is a consequence of mutations in the PEX7 gene, the GNPAT gene (which is located on chromosome 1) or the AGPS gene. The condition is acquired in an autosomal recessive manner.[2]
Pathophysiology
The mechanism of rhizomelic chondrodysplasia punctata in the case of type 1 of this condition one finds that peroxisome objective is PEX7, in peroxisome assembly. There are 3 pathways that count on PEX7 and are:[4][5]
- AGPS (catalyzes plasmalogen biosynthesis)
- PhYH (catalyzes catabolism of phytanic acid)
- ACAA1 (catalyzes beta-oxidation of VLCFA - straight)
Diagnosis
The diagnosis of rhizomelic chondrodysplasia punctata can be based on genetic testing[6] as well as radiography results, plus a physical examination of the individual.[3]
-
![Lower limbs/x-ray]]](./_assets_/ba48700f93e6ccab7a94adacb8f4c793/PMC4719397_JOCR-5-38-g005.png) Lower limbs/x-ray]]
Lower limbs/x-ray]] -
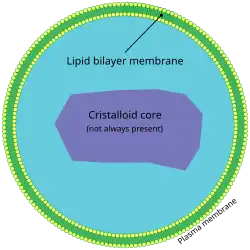 Peroxisome (this condition affects the peroxisome, causing peroxisome biogenesis disorders.)
Peroxisome (this condition affects the peroxisome, causing peroxisome biogenesis disorders.)
Types
- Type 1 (RCDP1) is associated with PEX7 mutations; these are peroxisome biogenesis disorders where proper assembly of peroxisomes is impaired.[4]
- Type 2 (RCDP2) is associated with DHAPAT mutations [7]
- Type 3 (RCDP3) is associated with AGPS mutations [8]
Treatment
Management of rhizomelic chondrodysplasia punctata can include physical therapy; additionally orthopedic procedures improved function sometimes in affected people.[4] However, the prognosis is poor in this condition.[3]
See also
- Plasmalogen
- Peroxisomal disorder
References
- ↑ 1.0 1.1 "Rhizomelic chondrodysplasia punctata type 1 | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Archived from the original on 24 January 2017. Retrieved 23 January 2017. Archived 24 January 2017 at the Wayback Machine
- ↑ 2.0 2.1 2.2 Reference, Genetics Home. "rhizomelic chondrodysplasia punctata". Genetics Home Reference. Archived from the original on 2016-12-24. Retrieved 2017-01-16. Archived 2016-12-24 at the Wayback Machine
- ↑ 3.0 3.1 3.2 RESERVED, INSERM US14 -- ALL RIGHTS. "Orphanet: Rhizomelic chondrodysplasia punctata". www.orpha.net. Archived from the original on 2 February 2017. Retrieved 23 January 2017.
{{cite web}}: CS1 maint: numeric names: authors list (link) Archived 2 February 2017 at the Wayback Machine - ↑ 4.0 4.1 4.2 4.3 4.4 Braverman, Nancy E.; Moser, Ann B.; Steinberg, Steven J. (1 January 1993). "Rhizomelic Chondrodysplasia Punctata Type 1". GeneReviews. PMID 20301447. Archived from the original on 18 January 2017. Retrieved 16 January 2017. Archived 18 January 2017 at the Wayback Machineupdate 2012
- ↑ Brodsky, Michael C. (2016-06-28). Pediatric Neuro-Ophthalmology. Springer. p. 620. ISBN 9781493933846. Archived from the original on 2021-08-29. Retrieved 2021-04-05.
- ↑ "Rhizomelic chondrodysplasia punctata type 1 - Conditions - GTR - NCBI". www.ncbi.nlm.nih.gov. Archived from the original on 8 February 2017. Retrieved 23 January 2017. Archived 8 February 2017 at the Wayback Machine
- ↑ "OMIM Entry - # 222765 - RHIZOMELIC CHONDRODYSPLASIA PUNCTATA, TYPE 2; RCDP2". omim.org. Archived from the original on 15 February 2020. Retrieved 16 January 2017. Archived 15 February 2020 at the Wayback Machine
- ↑ "OMIM Entry - # 600121 - RHIZOMELIC CHONDRODYSPLASIA PUNCTATA, TYPE 3; RCDP3". omim.org. Archived from the original on 2020-02-15. Retrieved 2017-01-16. Archived 2020-02-15 at the Wayback Machine
Further reading
- Benacerraf, Beryl (2007). Ultrasound of fetal syndromes (2nd ed.). Philadelphia: Churchill Livingstone / Elsevier. ISBN 978-0443066412. Archived from the original on 29 August 2021. Retrieved 23 January 2017.
- al.], [edited by] Kenneth F. Swaiman ... [et; Ashwal, Stephen; Ferriero, Donna M.; Schor, Nina F. (2012). Swaiman's pediatric neurology principles and practice (5th ed.). [Edinburgh]: Elsevier Saunders. ISBN 978-0323089111. Archived from the original on 29 August 2021. Retrieved 23 January 2017.
{{cite book}}:|first1=has generic name (help)
External links
| Classification | |
|---|---|
| External resources |
|